Five steps of tree of life generation
1) Gather data on species to be compared
Gather DNA information: For this project genome databases will be queried for DNA sequences
2) Align DNA sequences
Use string comparison algorithm to align DNA - String Edit for a O > (2^N) comparison series.
http://www.cs.princeton.edu/courses/archive/spr05/cos126/assignments/sequence.html
3) Build multitude of trees
Use distance attributes from string alignment to determine optimal solution.
4) Compare trees to obtain "true" tree
5) Perform post tree analysis
Performance Criteria
1) Run Time
2) Space Requirements
3) Statistical Performance
4) Topological Accuracy - How well the tree matches the true tree
5) Accuracy in relation to real world data
"A model is identifiable if it is uniquely characterized by the probability distribution it defines."
"A phylogenetic reconstruction method is statistically consistent under a model if the probability that the method reconstructs the true tree goes to 1 as the sequence length increases."
Markov models are identifiable.
"Phylogeny estimation typically is done under identifiable models."
• "Neighbour Joining is polynomial time, and statistically consistent."
• "Maximum Parsimony is NP-hard, and even exact solutions are not statistically consistent."
• "Maximum Likelihood is NP-hard, but exact solutions are statistically consistent"
"Neighbour Joining has poor performance on large diameter trees"
"Neighbor joining (and some other distance-based methods) will return the true tree with high probability provided sequence lengths are exponential in the diameter of the tree (Erdos et al., Atteson). Exponential lower bound for caterpillar trees: Lacey and Chang."
"Maximum likelihood will return the true tree with high probability provided sequence lengths are exponential in the number of taxa (Steel and Szekely)."
"Better accuracy is obtained through good heuristics for NP-hard optimization (esp. maximum likelihood)"
Saturday, 10 July 2010
Friday, 11 June 2010
Introduction to Phylogeny part 4
Still sourcing from Evolution 2nd edition.
"Alignment must be used to establish the homology of sequences"
Descendants inherit traits through genes. The history of descent is written in the DNA sequences. The molecular sequence is a simple string (character data). Characters are positions in the sequence, the character states are the nucleotides at those positions. This is a simplistic understanding of how to compare DNA, however it assumes that the dna strands are homologous, there are two problems with this:
1) with four nucleotides it is highly probably that two of the nucleotides will be the same just do to mutation.
2) In all but the most conserved genes, random bits of DNA have been inserted and deleted as the species have diverged from the common ancestor. This will cause orthologous gene sequences to differ in length, also nucleotides can be added or removed with in the sequence (base pair mutation).
To make the sequence homology consistent were a deletion has happened there needs to be a gap added and where there has been an insertion there must be a gap added to the other sequence. This is done so that the majority of the rest of the sequence should be homologous. Once the sequence is the same length we can compare nucleotides to scan for changes (mutations). The alignment is done based on assumptions about the ancestral sequence from which the derived sequence is though to descend. Thus alignment depends on assumptions about phylogeny and homology. Though algorithms exist to perform alignment automatically they can be unreliable and as such many alignments are done manually.
Even after a sequence has been align homoplasy is common (sequence that are similar for reasons other than recent shared ancestry). Homoplasy can be reduced but not elimiated by careful selection of character traits. This is one of the reasons that maximum likelihood is preferred as parsimony can be tricked by homoplasy.
"The neutral model and the molecular clock"
Dating of a clade starts with a fossil that approximates the divergence of the branches, many systemic methods assume that molecular mutation happens at about the same rate in each branch. The important part of this assumption is the regular rate of change in the nucleotides.
The fact that nucleotides change isn't controversial but the idea of the molecular clock (the idea that they mutate at the same rate) is controversial. However the idea of he molecular clock is important because it can help to predict the amount of time since the species diverged. The first problem with the nuclear clock is that though rates may be the same with in groups, they are different in other groups (mammals vs bacteria). Secondly generational time lapse is different between species (human vs bacteria). Third, the assumption is to "clock like", it would mean that each branch would have the same number of mutations and branches which is not the case. For these reasons many studies try to avoid using the molecular clock unless forced to do so by a lack of dated fossil evidence.
Using the right molecule for the right job
Because molecules decay at different rates using one suited for the time period you are measuring (uranium to measure things like the earth and moon, vs carbon for archaeological studies, mitochondrial DNA is useful for a few million years but not beyond, for deep time DNA analysis we need highly conserved DNA sequences which can go to 1500 million years).
The genealogy of genes can differ from the phylogeny of species.
But I think that is out of scope for this project.
"Alignment must be used to establish the homology of sequences"
Descendants inherit traits through genes. The history of descent is written in the DNA sequences. The molecular sequence is a simple string (character data). Characters are positions in the sequence, the character states are the nucleotides at those positions. This is a simplistic understanding of how to compare DNA, however it assumes that the dna strands are homologous, there are two problems with this:
1) with four nucleotides it is highly probably that two of the nucleotides will be the same just do to mutation.
2) In all but the most conserved genes, random bits of DNA have been inserted and deleted as the species have diverged from the common ancestor. This will cause orthologous gene sequences to differ in length, also nucleotides can be added or removed with in the sequence (base pair mutation).
To make the sequence homology consistent were a deletion has happened there needs to be a gap added and where there has been an insertion there must be a gap added to the other sequence. This is done so that the majority of the rest of the sequence should be homologous. Once the sequence is the same length we can compare nucleotides to scan for changes (mutations). The alignment is done based on assumptions about the ancestral sequence from which the derived sequence is though to descend. Thus alignment depends on assumptions about phylogeny and homology. Though algorithms exist to perform alignment automatically they can be unreliable and as such many alignments are done manually.
Even after a sequence has been align homoplasy is common (sequence that are similar for reasons other than recent shared ancestry). Homoplasy can be reduced but not elimiated by careful selection of character traits. This is one of the reasons that maximum likelihood is preferred as parsimony can be tricked by homoplasy.
"The neutral model and the molecular clock"
Dating of a clade starts with a fossil that approximates the divergence of the branches, many systemic methods assume that molecular mutation happens at about the same rate in each branch. The important part of this assumption is the regular rate of change in the nucleotides.
The fact that nucleotides change isn't controversial but the idea of the molecular clock (the idea that they mutate at the same rate) is controversial. However the idea of he molecular clock is important because it can help to predict the amount of time since the species diverged. The first problem with the nuclear clock is that though rates may be the same with in groups, they are different in other groups (mammals vs bacteria). Secondly generational time lapse is different between species (human vs bacteria). Third, the assumption is to "clock like", it would mean that each branch would have the same number of mutations and branches which is not the case. For these reasons many studies try to avoid using the molecular clock unless forced to do so by a lack of dated fossil evidence.
Using the right molecule for the right job
Because molecules decay at different rates using one suited for the time period you are measuring (uranium to measure things like the earth and moon, vs carbon for archaeological studies, mitochondrial DNA is useful for a few million years but not beyond, for deep time DNA analysis we need highly conserved DNA sequences which can go to 1500 million years).
The genealogy of genes can differ from the phylogeny of species.
But I think that is out of scope for this project.
Wednesday, 9 June 2010
Introduction to Phylogenetics part 3
For the same source as part 2, blogger had a fit at the size of the post.
Homology is the term used to describe a trait in two for more species derived from a common ancestor. Two morphological structures are called homologous if they are built by the same evolutionary pathway.
Orthology is the study of similarity in DNA sequences, two similar DNA sequences can be evidence of a recent common ancestor. Orthology works best when used on long DNA sequences as there is less chance that the sequence could have been derived naturally. Two similar DNA sequences are called orthologous.
Homology and orthology can be connected but it is not a reliable link. Evolution can re-task genes to new functions. When we find two species with similar DNA (orthology) but different morphological traits (homology) it is an indication of an "interesting" evolutionary event.
 Figure 1: an example of homologous characteristics deriving from a common ancestor.
Figure 1: an example of homologous characteristics deriving from a common ancestor.
Analogy describes similar characteristics that do not derive from a common ancestor.
 Figure 2: Analogous characteristics, as we can see the ichthyosaur and the dolphin look morphologically the same, however from the phylogenetic tree we can see that they do not share a common recent ancestor (we know that dolphins evolved from land mammals and mammals did not evolve from ichthyosaurs).
Figure 2: Analogous characteristics, as we can see the ichthyosaur and the dolphin look morphologically the same, however from the phylogenetic tree we can see that they do not share a common recent ancestor (we know that dolphins evolved from land mammals and mammals did not evolve from ichthyosaurs).
A broader term for analogy is homoplasy, a term to describe any similarity that does not derive from common ancestry.
How to build a phylogenetic tree
First you must collect specimens from the species that you want to place onto the phylogenetic tree. For each specimen you must determine what states particular traits are found, these traits are called characters.
Morphological traits are easier to track in samples that are more closely related (such as a bunch of butterflies) however if you are dealing with a diverse set of specimens (such as elephants and trees) then it is better to rely on DNA.
Next you build a matrix with the specimens on the rows and the characters on the columns. The characters selected should be meaningful and useful in defining a relationship. Systematics teaches us that only shared derived traits are useful for classification.
The ingroup is the collection of species that you are focused on sharing a derived character.
The outgroup is the relatives to the ingroup but do not share the character in question.
So derived characters shared by the ingroup but not shared with the outgroup are informative, primitive characters (from the ancient past) are less informative. Therefore it is important to know which characters are derived and which are primitive. Which leads to the problem of you cannot determine a derived characteristic from a primitive one with out a tree and you cannot have a tree with out knowing if a character is derived or primitive.
Solutions to the problem:
Parsimony: Build all versions of the tree and select the simplest using Occam's Razor to select the tree with the least convergence and fewest changes in character state.
Maximum Likelihood: Choose the tree that would make the characters you did observe most likely. This method requires an assumption of how evolutionary change happens over time.
Bayesian inference: Choose the tree that is most likely (branches, branch lengths, and character distributions) given a prior expectation of what the tree should look like. This method uses statistics and probability to determine the correct tree.
Hybrid: Use a combination of the three listed solutions.
A deeper look into phylogenetics:
Synapomorphy: "A shared character state indicating that two species belong to the same group." A trait derived from a common ancestor such as the forelimbs in tetrapods and not found in lobed fish.
Symplesiomorphy: an uninformative ancestral shared trait.
Some important points:
1) Looking the same is not informative.
2) Informative traits are shared and derived.
3) What is shared and what is derived depends on the context of what part of the tree you are looking at. Therefore what is informative is dependent on context.
To build the tree from the character matrix follow the change in character states between species
"We start to build a phylogenetic tree by considering the elemental step—the
change of a trait from one state to another (Figure 13.16). We denote the things
whose relationships are being analyzed—the genes, species, or larger groups—
by capital letters, A, B, C . . ., and the traits being used to determine relationship
by numbers, 1, 2, 3 . . .."
How do we infer the correct tree from the character matrix? We start with the observable character matrix and build the tree from it.
 Figure 3: an example of how a character matrix relates and is transformed into a phylogenetic tree.
Figure 3: an example of how a character matrix relates and is transformed into a phylogenetic tree.
Trees are rooted by choice of outgroup.
A tree can be generated by focusing on the synapomorphy or the symplesiomorphy as the basis for branches. Once the trees have been made we can use parsimony or maximum likelihood to choose which one we think is correct. After the tree is chosen we must determine the root, that is done by choosing an outgroup that is closely related to the clade. So if we are establishing a clade of mammals then using a fish or a lizard as the outgroup would be appropriate to establish an ancestor relationship. "Rooting the tree establishes the direction of character change within the ingroup." The character states in the outgroup are considered to be primitive.
Outgroup comparision: A method used to root a phylogenetic tree and to establish the direction of character change.
 Figure 4: Rooting a phylogenetic tree
Figure 4: Rooting a phylogenetic tree
 Figure 5: Selecting a tree using parsimony, the tree with the least amount of step changes is more likely to be correct.
Figure 5: Selecting a tree using parsimony, the tree with the least amount of step changes is more likely to be correct.
Using maximum likelihood means that we need a way to determine the probability that the tree is correct. This is most likely a model based on the probabilities that a change in character state was random. "For each candidate tree the probability is calculated of finding any two
sequences at opposite ends of a branch (for example a T at one end and a C at
the other). This is done for all branches, the probabilities are multiplied together
to get the likelihood for the whole tree, and the best tree is then the one with the
maximum likelihood (Felsenstein 1988). The method is logically appealing and
computationally expensive. Its range of application is increasing as computers
improve."
Constructing a even a modest tree requires immense computational power.
Hence the reason for this project.
Though a tree is built from a simple matrix, as species and character traits are added the problem becomes immense (see figure 6, it is greater than O (2^n))
 Figure 6: Computational estimates of various sized phylogenetic trees. (Note to the reader: there are only around 10^130 protons, electrons and neutrons in the universe, compare that to the number of possible trees for 500 taxa which is around 10^1280 possible trees, no wonder creative computer algorithms are necessary! Given a computer running at a teraflop (10^13 instructions per second) it would take 10^1267 seconds to process it and the universe is only about 4 x 10^17 seconds old so it would take about 10^1250 lifetimes of the universe to compute! That amount of time is so big that I can't get my head around it, it may as well be infinite, but thankfully I can think of some algorithms to bring that number down, we shall have to see if they work ;) but even the ones that leap to mind would still be limited to about 25 species.)
Figure 6: Computational estimates of various sized phylogenetic trees. (Note to the reader: there are only around 10^130 protons, electrons and neutrons in the universe, compare that to the number of possible trees for 500 taxa which is around 10^1280 possible trees, no wonder creative computer algorithms are necessary! Given a computer running at a teraflop (10^13 instructions per second) it would take 10^1267 seconds to process it and the universe is only about 4 x 10^17 seconds old so it would take about 10^1250 lifetimes of the universe to compute! That amount of time is so big that I can't get my head around it, it may as well be infinite, but thankfully I can think of some algorithms to bring that number down, we shall have to see if they work ;) but even the ones that leap to mind would still be limited to about 25 species.)
That is probably a good place to say: To be continued in part 4
Homology is the term used to describe a trait in two for more species derived from a common ancestor. Two morphological structures are called homologous if they are built by the same evolutionary pathway.
Orthology is the study of similarity in DNA sequences, two similar DNA sequences can be evidence of a recent common ancestor. Orthology works best when used on long DNA sequences as there is less chance that the sequence could have been derived naturally. Two similar DNA sequences are called orthologous.
Homology and orthology can be connected but it is not a reliable link. Evolution can re-task genes to new functions. When we find two species with similar DNA (orthology) but different morphological traits (homology) it is an indication of an "interesting" evolutionary event.
 Figure 1: an example of homologous characteristics deriving from a common ancestor.
Figure 1: an example of homologous characteristics deriving from a common ancestor.Analogy describes similar characteristics that do not derive from a common ancestor.
 Figure 2: Analogous characteristics, as we can see the ichthyosaur and the dolphin look morphologically the same, however from the phylogenetic tree we can see that they do not share a common recent ancestor (we know that dolphins evolved from land mammals and mammals did not evolve from ichthyosaurs).
Figure 2: Analogous characteristics, as we can see the ichthyosaur and the dolphin look morphologically the same, however from the phylogenetic tree we can see that they do not share a common recent ancestor (we know that dolphins evolved from land mammals and mammals did not evolve from ichthyosaurs).A broader term for analogy is homoplasy, a term to describe any similarity that does not derive from common ancestry.
How to build a phylogenetic tree
First you must collect specimens from the species that you want to place onto the phylogenetic tree. For each specimen you must determine what states particular traits are found, these traits are called characters.
Morphological traits are easier to track in samples that are more closely related (such as a bunch of butterflies) however if you are dealing with a diverse set of specimens (such as elephants and trees) then it is better to rely on DNA.
Next you build a matrix with the specimens on the rows and the characters on the columns. The characters selected should be meaningful and useful in defining a relationship. Systematics teaches us that only shared derived traits are useful for classification.
The ingroup is the collection of species that you are focused on sharing a derived character.
The outgroup is the relatives to the ingroup but do not share the character in question.
So derived characters shared by the ingroup but not shared with the outgroup are informative, primitive characters (from the ancient past) are less informative. Therefore it is important to know which characters are derived and which are primitive. Which leads to the problem of you cannot determine a derived characteristic from a primitive one with out a tree and you cannot have a tree with out knowing if a character is derived or primitive.
Solutions to the problem:
Parsimony: Build all versions of the tree and select the simplest using Occam's Razor to select the tree with the least convergence and fewest changes in character state.
Maximum Likelihood: Choose the tree that would make the characters you did observe most likely. This method requires an assumption of how evolutionary change happens over time.
Bayesian inference: Choose the tree that is most likely (branches, branch lengths, and character distributions) given a prior expectation of what the tree should look like. This method uses statistics and probability to determine the correct tree.
Hybrid: Use a combination of the three listed solutions.
A deeper look into phylogenetics:
Synapomorphy: "A shared character state indicating that two species belong to the same group." A trait derived from a common ancestor such as the forelimbs in tetrapods and not found in lobed fish.
Symplesiomorphy: an uninformative ancestral shared trait.
Some important points:
1) Looking the same is not informative.
2) Informative traits are shared and derived.
3) What is shared and what is derived depends on the context of what part of the tree you are looking at. Therefore what is informative is dependent on context.
To build the tree from the character matrix follow the change in character states between species
"We start to build a phylogenetic tree by considering the elemental step—the
change of a trait from one state to another (Figure 13.16). We denote the things
whose relationships are being analyzed—the genes, species, or larger groups—
by capital letters, A, B, C . . ., and the traits being used to determine relationship
by numbers, 1, 2, 3 . . .."
How do we infer the correct tree from the character matrix? We start with the observable character matrix and build the tree from it.
 Figure 3: an example of how a character matrix relates and is transformed into a phylogenetic tree.
Figure 3: an example of how a character matrix relates and is transformed into a phylogenetic tree.Trees are rooted by choice of outgroup.
A tree can be generated by focusing on the synapomorphy or the symplesiomorphy as the basis for branches. Once the trees have been made we can use parsimony or maximum likelihood to choose which one we think is correct. After the tree is chosen we must determine the root, that is done by choosing an outgroup that is closely related to the clade. So if we are establishing a clade of mammals then using a fish or a lizard as the outgroup would be appropriate to establish an ancestor relationship. "Rooting the tree establishes the direction of character change within the ingroup." The character states in the outgroup are considered to be primitive.
Outgroup comparision: A method used to root a phylogenetic tree and to establish the direction of character change.
 Figure 4: Rooting a phylogenetic tree
Figure 4: Rooting a phylogenetic tree Figure 5: Selecting a tree using parsimony, the tree with the least amount of step changes is more likely to be correct.
Figure 5: Selecting a tree using parsimony, the tree with the least amount of step changes is more likely to be correct.Using maximum likelihood means that we need a way to determine the probability that the tree is correct. This is most likely a model based on the probabilities that a change in character state was random. "For each candidate tree the probability is calculated of finding any two
sequences at opposite ends of a branch (for example a T at one end and a C at
the other). This is done for all branches, the probabilities are multiplied together
to get the likelihood for the whole tree, and the best tree is then the one with the
maximum likelihood (Felsenstein 1988). The method is logically appealing and
computationally expensive. Its range of application is increasing as computers
improve."
Constructing a even a modest tree requires immense computational power.
Hence the reason for this project.
Though a tree is built from a simple matrix, as species and character traits are added the problem becomes immense (see figure 6, it is greater than O (2^n))
 Figure 6: Computational estimates of various sized phylogenetic trees. (Note to the reader: there are only around 10^130 protons, electrons and neutrons in the universe, compare that to the number of possible trees for 500 taxa which is around 10^1280 possible trees, no wonder creative computer algorithms are necessary! Given a computer running at a teraflop (10^13 instructions per second) it would take 10^1267 seconds to process it and the universe is only about 4 x 10^17 seconds old so it would take about 10^1250 lifetimes of the universe to compute! That amount of time is so big that I can't get my head around it, it may as well be infinite, but thankfully I can think of some algorithms to bring that number down, we shall have to see if they work ;) but even the ones that leap to mind would still be limited to about 25 species.)
Figure 6: Computational estimates of various sized phylogenetic trees. (Note to the reader: there are only around 10^130 protons, electrons and neutrons in the universe, compare that to the number of possible trees for 500 taxa which is around 10^1280 possible trees, no wonder creative computer algorithms are necessary! Given a computer running at a teraflop (10^13 instructions per second) it would take 10^1267 seconds to process it and the universe is only about 4 x 10^17 seconds old so it would take about 10^1250 lifetimes of the universe to compute! That amount of time is so big that I can't get my head around it, it may as well be infinite, but thankfully I can think of some algorithms to bring that number down, we shall have to see if they work ;) but even the ones that leap to mind would still be limited to about 25 species.)That is probably a good place to say: To be continued in part 4
Introduction to Phylogenetics part 2
From the Stearns and Hoekstra book Evolution 2nd edition, chapter 13 Phylogenetics and systemantics
Phylogeny refers to the history and relations of organisms.
From the Greek phyl- meaning tribe and gen- origin or descent.
The goal of phylogeny is to map the relationships of all organisms that we have evidence to have existed.
The phylogenetic tree has three main branches: Bacteria, Archaea, and Eukaryota
Bacteria: single-celled or noncellular spherical or spiral or rod-shaped organisms lacking chlorophyll that reproduce by fission; important as pathogens and for biochemical properties; taxonomy is difficult; often considered to be plants Ref: Princeton
Archaea: Single-celled organisms similar to bacteria as they do not have their genetic material enclosed within a nucleus. Archaea are prokaryotic but genetically similar to eukaryotes so are placed in a separate kingdom 'the archaea'. Ref: Australian Academy of Science
Eukaryota: The domain comprised of eukaryotes or organisms whose cells contain a true nucleus. Ref: Biology Online
Any complete branch of the phylogenetic tree is called a clade. A group of organisms that share a recent common ancestor.
At the largest scale the phylogenetic tree is dominated by bacteria and the phylogenetic tree spans 3.7 billion years. From mitochondrial eve to present day organisms.
 Figure 1: From Darwin's Origin of Species (1859) The lowest ancestors A, B, C, D, E and descendants. Only A and E have descendants that reach the time of study.
Figure 1: From Darwin's Origin of Species (1859) The lowest ancestors A, B, C, D, E and descendants. Only A and E have descendants that reach the time of study.
 Figure 2: From Evolution 2nd edition. A more complete phylogenetic tree.
Figure 2: From Evolution 2nd edition. A more complete phylogenetic tree.
It should be noted that the ability for Eukaryotes gained the ability to use chlorophyll and breathe comes from a symbiotic relationship with Bacteria.
The vast majority of the phylogenetic tree is dedicated to single cell organisms
There are three main types of multicellular organisms: Plants, Fungi, and Animals. They share a common ancestor approximately .8-1 billion years ago.
 Figure 3: The phylogenetic tree for multicellular organisms from about 800 million years ago to present day. Source Evolution 2nd edition
Figure 3: The phylogenetic tree for multicellular organisms from about 800 million years ago to present day. Source Evolution 2nd edition
Phylogenetic relationships are not obvious, they must be discovered. Related organisms are similar in important ways and the more distantly related organisms are different in other important ways.
Early biologists could only use morphological data to determine how organisms are related but now biologists can use molecular traits to quickly identify relationships through DNA inheritance. Using DNA can specify relationships back in time allowing for correct ancestor relationships.
Some modern phylogenetic relationships are surprising, such as the fact that the closest relative to a whale is a hippopotamus. Since whales are highly adapted to life in water using morphological data is difficult to classify them in relation to land animals so molecular data must be used.

Figure 4: some of the surprising relationships illuminated by modern phylogenetics. Source Evolution 2nd edition.
Another surprise is that giant pandas and lesser pandas are not closely related. The giant panda is related to bears and the lesser panda is related to weasels and raccoons.
Monophyletic groups are a group where the most recent common ancestor is not the ancestor of any other group. The dog clade is an example of a monophyletic group.
A paraphyletic group is an unnatural group that does not include all the descendants of the most recent common ancestor. Lizards are a good examle, since the lizard group does not include birds or mammals that also descend from the most recent common ancestor.
A polyphyletic group is one that descends from a number of ancestors. Worms are a good example of a polyphyletic group as the different species (flat worms, earth worms) descend from different ancestors.
 Figure 5: An example of a monophyletic group versus a paraphyletic group, source Evolution 2nd edition
Figure 5: An example of a monophyletic group versus a paraphyletic group, source Evolution 2nd edition
Adaptive convergence is where differing species adapt to similar conditions gaining similar morphological traits, such as a dolphin and a tuna having similar slimline shapes in order to swim faster than a non-slimline shape without having a recent common ancestor. Adaptive convergence can confuse phylogenetic taxonomy when species are grouped by morphological data.
Divergence is the opposite of convergence, it is when a species adapts in a new way to a new or changing environment in a different way to close relatives. This is the cause of speciation, but it can also confuse phylogenetic taxonomy as some species have wildly divergent morphology while still being the same species.
Taken together convergence and divergence can really confuse the attempt to build a phylogenetic tree as shared traits (morphological and molecular) must be traced to a common ancestor.
To be continued in part 3
Phylogeny refers to the history and relations of organisms.
From the Greek phyl- meaning tribe and gen- origin or descent.
The goal of phylogeny is to map the relationships of all organisms that we have evidence to have existed.
The phylogenetic tree has three main branches: Bacteria, Archaea, and Eukaryota
Bacteria: single-celled or noncellular spherical or spiral or rod-shaped organisms lacking chlorophyll that reproduce by fission; important as pathogens and for biochemical properties; taxonomy is difficult; often considered to be plants Ref: Princeton
Archaea: Single-celled organisms similar to bacteria as they do not have their genetic material enclosed within a nucleus. Archaea are prokaryotic but genetically similar to eukaryotes so are placed in a separate kingdom 'the archaea'. Ref: Australian Academy of Science
Eukaryota: The domain comprised of eukaryotes or organisms whose cells contain a true nucleus. Ref: Biology Online
Any complete branch of the phylogenetic tree is called a clade. A group of organisms that share a recent common ancestor.
At the largest scale the phylogenetic tree is dominated by bacteria and the phylogenetic tree spans 3.7 billion years. From mitochondrial eve to present day organisms.
 Figure 1: From Darwin's Origin of Species (1859) The lowest ancestors A, B, C, D, E and descendants. Only A and E have descendants that reach the time of study.
Figure 1: From Darwin's Origin of Species (1859) The lowest ancestors A, B, C, D, E and descendants. Only A and E have descendants that reach the time of study. Figure 2: From Evolution 2nd edition. A more complete phylogenetic tree.
Figure 2: From Evolution 2nd edition. A more complete phylogenetic tree.It should be noted that the ability for Eukaryotes gained the ability to use chlorophyll and breathe comes from a symbiotic relationship with Bacteria.
The vast majority of the phylogenetic tree is dedicated to single cell organisms
There are three main types of multicellular organisms: Plants, Fungi, and Animals. They share a common ancestor approximately .8-1 billion years ago.
 Figure 3: The phylogenetic tree for multicellular organisms from about 800 million years ago to present day. Source Evolution 2nd edition
Figure 3: The phylogenetic tree for multicellular organisms from about 800 million years ago to present day. Source Evolution 2nd editionPhylogenetic relationships are not obvious, they must be discovered. Related organisms are similar in important ways and the more distantly related organisms are different in other important ways.
Early biologists could only use morphological data to determine how organisms are related but now biologists can use molecular traits to quickly identify relationships through DNA inheritance. Using DNA can specify relationships back in time allowing for correct ancestor relationships.
Some modern phylogenetic relationships are surprising, such as the fact that the closest relative to a whale is a hippopotamus. Since whales are highly adapted to life in water using morphological data is difficult to classify them in relation to land animals so molecular data must be used.

Figure 4: some of the surprising relationships illuminated by modern phylogenetics. Source Evolution 2nd edition.
Another surprise is that giant pandas and lesser pandas are not closely related. The giant panda is related to bears and the lesser panda is related to weasels and raccoons.
Monophyletic groups are a group where the most recent common ancestor is not the ancestor of any other group. The dog clade is an example of a monophyletic group.
A paraphyletic group is an unnatural group that does not include all the descendants of the most recent common ancestor. Lizards are a good examle, since the lizard group does not include birds or mammals that also descend from the most recent common ancestor.
A polyphyletic group is one that descends from a number of ancestors. Worms are a good example of a polyphyletic group as the different species (flat worms, earth worms) descend from different ancestors.
 Figure 5: An example of a monophyletic group versus a paraphyletic group, source Evolution 2nd edition
Figure 5: An example of a monophyletic group versus a paraphyletic group, source Evolution 2nd editionAdaptive convergence is where differing species adapt to similar conditions gaining similar morphological traits, such as a dolphin and a tuna having similar slimline shapes in order to swim faster than a non-slimline shape without having a recent common ancestor. Adaptive convergence can confuse phylogenetic taxonomy when species are grouped by morphological data.
Divergence is the opposite of convergence, it is when a species adapts in a new way to a new or changing environment in a different way to close relatives. This is the cause of speciation, but it can also confuse phylogenetic taxonomy as some species have wildly divergent morphology while still being the same species.
Taken together convergence and divergence can really confuse the attempt to build a phylogenetic tree as shared traits (morphological and molecular) must be traced to a common ancestor.
To be continued in part 3
Tuesday, 8 June 2010
Introduction to Phylogenetics
Phylogenetics is the science of Phylogeny, and as such I will be focusing on it instead of the more generic Phylogeny for this post. I have found this article as a starting point.
The goal of Phylogeny is to construction a phylogenetic tree that maps the genetic relationships of all species (past and present) on earth.
Phylogeny uses evolutionary distance/evolutionary relationships to determine the taxonomy of species.
Taxonomy is the science of naming, classifying and describing species.
Taxonomy groups organisms into taxa (groups)
Taxa are then further grouped by biological similarity.
Systematics adds to taxonomy and allows for taxa to be grouped into species, based on evolutionary similarities and mechanisms for evolution.
William Hennig (1950s) said systematics should follow known evolutionary lineage, thereby creating the field of phylogenetic semantics.
Phylogenetic systematics deals with the classification of organisms based on evolutionary relationships.
Phylogenetic relationships used to be determined by morphological data, this was found to be insufficient.
Cladistics was the field that developed to determine phylogentic relationships based on shared, derived characteristics.
There are two types of characteristics: primitive traits and derived traits.
Primitive traits are those inherited from the ancestor of the group. Such as all monkeys have tails so having a tail is a primitive trait for a monkey.
Derived traits are those that have evolved in the group under study. Size, colouring, behaviour and diet are derived traits (as I understand it). Birds are an excellent example of derived traits like the differences between a hummingbird and an eagle.
The simple explanation of the source of a derived trait is that it is from a evolutionarily more recent ancestor than the earlier ancestor being examined.
There are two main types of analysis to determine phylogenetic relationships: Phenetic and Cladistic.
Phenetic methodology (numerical taxonomy) uses over all similarities to classify relationships, once similarities are determined the organisms are grouped into clusters called phenograms.
Cladistic methodology uses the idea that members of a group share a common ancestry and are more closely related to each other than any other organism, so my children are in the same group (siblings) since they are one step removed from me as an ancestor. The shared derived characteristics in the group are called synapomorphies.
There are two recent tools that have helped phylogenetics: computer algorithms that can automatically place groups into a phylogenetic tree and the ability of using molecular sequence data (DNA?) for phylogenetic studies.
Phylogenetics can use both molecular and morphological data to classify organisms.
Molecular studies use genes as the basis of comparison. The assumption is that organisms that share genes will be more closely related than those that don't.
Morphological studies use an organism's phenotype to determine relationships.
phenotype ( ) n. The observable physical or biochemical characteristics of an organism, as determined by both genetic makeup (sic) and environmental. according to Answers.com
Both methods are related as the genome is used in both methodologies and the genome has a strong influence in the phenotype. The molecular methodology has an advantage in that it can be studied with out morphological expressions.
Closely related species have a more recent ancestor than a distantly related species (two different types of hummingbird versus an eagle) and those relationships can be expressed by a phylogenetic tree.
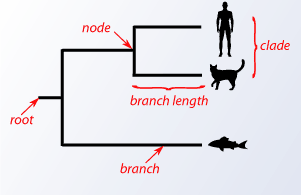
A phylogenetic tree is a graphical representation of how species are interrelated and have nodes and branches. A node is a taxonomical species and the branch is how it is related to another taxonomical species with the length of the branch reprisenting the amount of evolutionary distance between taxonomical species.
There are two main types of tree construction methods: Cladastic (maximum parsimony and maximum likelyhood) and Phenetic (distance matrix method).
picture of a simple phylogenetic tree
Maximum parsimony asserts that Occam's razor should be applied and that simple hypotheses are preferable to complex ones, the minimum number of genetic changes should be used to show phylogenetic relationships, the procedure compares a number of parsimony trees and chooses the simplest one.
Maximum likelihood examines a number of possible trees and chooses the best one based on some criteria. This approach takes into concideration the evolutionary process that can change one gene into another.
Distance matrix examines the mean changes in the genetics of a pair of taxa to determine the distance relationship between the two taxa. Gene sequences must be simplified in order for them to be compared and the difference in the two genomes can be interrupted as the distance between the two taxonomical species.
Molecular phylogeny was first proposed by Pauling and Zuckerkandl (1962) when they noted the rates at which amino acids were substituted in animal hemoglobin at a predictable rate over time. This is advantageous as genotypes can be studied directly and organisms can be compared directly even if they are morphologically very different, as such this method does not rely on phenotype.
Biologist calculate that there are between 5 and 10 million species alive today and genetic information indicates that all of them may have a common ancestor, hence the reason to study the Tree of Life to determine the root of all life on the planet.
Monday, 7 June 2010
The basics of parallel computing
From here
Basics of parallelism:
To understand parallelism, first you must understand serialism. A serial process is one that starts from the beginning and follows its instructions until the end. A parallel process breaks up the instructions and spreads them out onto multiple processing units and thereby speeds up the execution of the process by adding cost to the equation, both costs in equipment and in parallel overhead (the extra control functions that manage the process). A parallel process will often (most of the time) include serial sections of instructions.
A simple way of understanding a serial process is to think of a librarian putting away 100 books that have just been returned to the library. If the time it takes to shelve one book is called one unit of time then it will take one librarian 100 units of time to shelve all the books.
To turn this into a parallel process we add 9 additional librarians and split the 100 books between the 10 librarians each librarian will be required to shelve 10 books, taking 10 units of time each (concurrently so it only takes 10 units of real time). But there is some over head in additional salaries and the time it takes to split the books between the librarians and for the librarians to report that the task is done. So it takes a little more than 10 units of time for the 10 librarians to perform the same task as the single librarian.
What is speed up and how do we measure it?
Speed up is the factor that a process in sped up by making it parallel. The equation is fairly simple: (old speed)/(new speed).
So for our librarian problem the old speed is 100 and the new speed is 10 (ignoring parallel overhead)
100/10 = 10 so the speed up of adding 9 librarians is 10.
Are there limits to what the speed up can be?
Yes, there are hardware limitations on how fast a process can be spread and there is a level at which adding more parallel capability is redundant.
If we increase the number of librarians to 100 we still see a speed up as the total time taken by the librarians will be 1
100/1 = 100 speed up
However is we have 1000 librarians we will see no additional speed up as the time taken will still be 1
100/1 = 100 speed up
So in this instance the additional 900 librarians will be sitting idle, however an additional 900 books could be added to the processing queue with out the process taking any additional time.
What kinds of parallel computing are there?
There are 2 variables with 2 values to concider according to Flynn's Classical Taxonomy.
Number of instructions: single or many
Data: single or many
So the four types of parallel computing are:
Single Instruction, Single Data: In this senario a single set of instructions are processed on the same data on multiple cores.
Single Instruction, Multiple Data: This senario has the same set of instructions workingon more than one set of data
Multiple Instruction, Single Data: This senario has multiple instructions working on a single dataset.
Multiple Instruction, Multiple Data: This senario has multiple processors executing instructions on different data sets. This is the mostly likely form of parallel processing for this project.
Important terms:
Task: A series of instructions
Parallel Task: A series of instructions that are safe to run in parallel.
Serial Execution: A set of instructions that must be runone after another.
Parallel Execution: A set of instructions that can be run on more than one processor at a time.
Pipelining: The process by which a set of instructions is broken up into discrete parts in order to be processed in parallel.
Shared Memory: Memory shared by all parallel processes.
Symetric Multiprocessing (SMP): A form of parallelism that multiple processors share the same address and memory and work on the same problem at the same time (using shared memory).
Distributed Memory: Memory spread accross a network. A process must use the network to see any non-local memory.
Communications: Typically a process will have to communicate with other proceses, also known as Inter Process Communication (IPC)
Syncronization: The process by with some threads are delayed in order for others to catch up (end of process or for communication)
Granularity: How much process is going on vs how much communication is going on.
Scalability: How the process responds to additional processor resources.
Basics of parallelism:
To understand parallelism, first you must understand serialism. A serial process is one that starts from the beginning and follows its instructions until the end. A parallel process breaks up the instructions and spreads them out onto multiple processing units and thereby speeds up the execution of the process by adding cost to the equation, both costs in equipment and in parallel overhead (the extra control functions that manage the process). A parallel process will often (most of the time) include serial sections of instructions.
A simple way of understanding a serial process is to think of a librarian putting away 100 books that have just been returned to the library. If the time it takes to shelve one book is called one unit of time then it will take one librarian 100 units of time to shelve all the books.
To turn this into a parallel process we add 9 additional librarians and split the 100 books between the 10 librarians each librarian will be required to shelve 10 books, taking 10 units of time each (concurrently so it only takes 10 units of real time). But there is some over head in additional salaries and the time it takes to split the books between the librarians and for the librarians to report that the task is done. So it takes a little more than 10 units of time for the 10 librarians to perform the same task as the single librarian.
What is speed up and how do we measure it?
Speed up is the factor that a process in sped up by making it parallel. The equation is fairly simple: (old speed)/(new speed).
So for our librarian problem the old speed is 100 and the new speed is 10 (ignoring parallel overhead)
100/10 = 10 so the speed up of adding 9 librarians is 10.
Are there limits to what the speed up can be?
Yes, there are hardware limitations on how fast a process can be spread and there is a level at which adding more parallel capability is redundant.
If we increase the number of librarians to 100 we still see a speed up as the total time taken by the librarians will be 1
100/1 = 100 speed up
However is we have 1000 librarians we will see no additional speed up as the time taken will still be 1
100/1 = 100 speed up
So in this instance the additional 900 librarians will be sitting idle, however an additional 900 books could be added to the processing queue with out the process taking any additional time.
What kinds of parallel computing are there?
There are 2 variables with 2 values to concider according to Flynn's Classical Taxonomy.
Number of instructions: single or many
Data: single or many
So the four types of parallel computing are:
Single Instruction, Single Data: In this senario a single set of instructions are processed on the same data on multiple cores.
Single Instruction, Multiple Data: This senario has the same set of instructions workingon more than one set of data
Multiple Instruction, Single Data: This senario has multiple instructions working on a single dataset.
Multiple Instruction, Multiple Data: This senario has multiple processors executing instructions on different data sets. This is the mostly likely form of parallel processing for this project.
Important terms:
Task: A series of instructions
Parallel Task: A series of instructions that are safe to run in parallel.
Serial Execution: A set of instructions that must be runone after another.
Parallel Execution: A set of instructions that can be run on more than one processor at a time.
Pipelining: The process by which a set of instructions is broken up into discrete parts in order to be processed in parallel.
Shared Memory: Memory shared by all parallel processes.
Symetric Multiprocessing (SMP): A form of parallelism that multiple processors share the same address and memory and work on the same problem at the same time (using shared memory).
Distributed Memory: Memory spread accross a network. A process must use the network to see any non-local memory.
Communications: Typically a process will have to communicate with other proceses, also known as Inter Process Communication (IPC)
Syncronization: The process by with some threads are delayed in order for others to catch up (end of process or for communication)
Granularity: How much process is going on vs how much communication is going on.
Scalability: How the process responds to additional processor resources.
Definitions
I have just been given my final year project: Parallel Alorithms for Phylogeny.
What are parallel algorithms?
A parallel algorithm is one that breaks a larger problem into distinct subproblems and then solves them using an array of processing units instead of doing the whole problem on one CPU.
For more information
What is Phylogeny?
Phylogeny is the study of how species relate to each other over time. It is related to paleontology and taxonomy (the grouping of individuals into species and species in to genus, etc). The science of phylogeny is called phylogenetics.
For more information
What is my goal?
To build a fast parallel algorithm to determine the phylogenetic relation between miltiple sets of DNA to determine how closely related they are and to hopefully be able to determine how long ago they split from a common ancestor.
What are parallel algorithms?
A parallel algorithm is one that breaks a larger problem into distinct subproblems and then solves them using an array of processing units instead of doing the whole problem on one CPU.
For more information
What is Phylogeny?
Phylogeny is the study of how species relate to each other over time. It is related to paleontology and taxonomy (the grouping of individuals into species and species in to genus, etc). The science of phylogeny is called phylogenetics.
For more information
What is my goal?
To build a fast parallel algorithm to determine the phylogenetic relation between miltiple sets of DNA to determine how closely related they are and to hopefully be able to determine how long ago they split from a common ancestor.
Subscribe to:
Comments (Atom)
